Sequence comparison is a crucial aspect of bioinformatics analysis that involves comparing newly determined biological sequences with previously known sequences stored in databases.
Sequence alignment is considered the most essential step in comparing biological sequences. Sequence alignment arranges two or more nucleotide or amino acid sequences to identify regions of similarity between the sequences. These regions of similarity are helpful in understanding the functional, structural, and evolutionary relationships between the sequences.
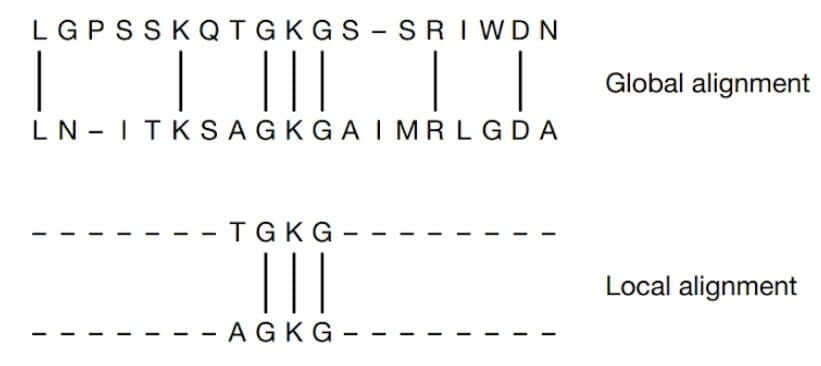
Two commonly used sequence alignment algorithms are global alignment and local alignment.
Global alignment: Global alignment is a method of comparing two sequences, which aligns the entire length of the sequences by maximizing the overall similarity. This method is used when comparing sequences that are of the same length.
Local alignment: In local alignment, instead of attempting to align the entire length of the sequences, only the regions with the highest density of matches are aligned. This is useful for identifying short conserved regions in protein or nucleotide sequences.
Interesting Science Videos
Types of Sequence Alignment
A. Pairwise Alignment
- Pairwise sequence alignment is the type of sequence alignment that involves aligning two sequences to identify the optimal pairing of the sequences.
- It is based on a scoring system that assigns positive scores to matching characters and negative scores to mismatching characters or gaps.
- The main objective of pairwise sequence alignment is to obtain the highest possible score, which indicates the degree of similarity between the two sequences.
B. Multiple Sequence Alignment
- Multiple Sequence Alignment involves aligning multiple (three or more) biological sequences to achieve optimal sequence matching.
- Multiple sequence alignments are used to identify conserved sequence regions and to construct phylogenetic trees, which help us understand the functional and evolutionary relationships between different species or groups of organisms.
Methods of pairwise sequence alignment
There are three main methods for generating pairwise alignments:
A. Dot-matrix method
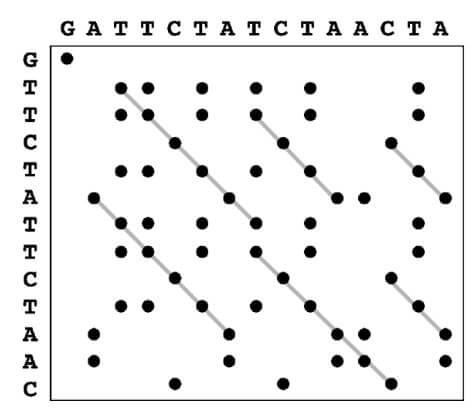
- Dot matrix method, also known as the dot plot method, is a graphical method of sequence alignment that involves comparing two sequences by plotting them in a two-dimensional matrix.
- In a dot matrix, two sequences that must be compared are plotted along a matrix’s horizontal and vertical axes. The method then scans each residue of one sequence to identify similarities with all residues in the other sequence.
- If a residue in one sequence matches a residue in the other sequence, a dot is placed in the corresponding position in the matrix. Otherwise, the matrix position is left blank.
- If the two sequences being compared are highly similar, the dot plot will display as a single line along the matrix’s main diagonal. However, when the sequences are less similar, the dot plot will show more scattered dots with fewer diagonal lines, indicating that the sequences share less similarity.
- Dot plots can also find repeat elements in a single sequence. Short parallel lines above and below the main diagonal indicate the presence of repeats.
B. Dynamic programming
- Dynamic programming is used to find the optimal alignment between two proteins or nucleic acid sequences by comparing all possible pairs of characters in the sequences.
- Dynamic programming can be used to produce both global and local alignments. The global pairwise alignment algorithm using dynamic programming is based on the Needleman-Wunsch algorithm, while the dynamic programming in local alignment is based on the Smith-Waterman algorithm.
This method works in the following three steps.
- Initialization of the scoring matrix: The first step is to create a two-dimensional matrix where the two sequences to be aligned are written along the top and left sides. The matrix is initialized with gap penalties and an initial score of zero at the top-left corner.
- Matrix filling with maximum scores: The next step involves filling the matrix with scores based on a scoring matrix. Scoring matrices for nucleotide sequences are simple. A positive value is given for a match, and a negative value for a mismatch. For amino acids, BLOSUM and PAM scoring matrices are used.
To calculate the alignment scores, the algorithm starts at the upper left corner of the matrix and proceeds one row at a time toward the lower right corner. The algorithm fills each cell in the matrix with the maximum score that can be obtained by aligning the corresponding residues. - Traceback to identify optimal alignment: After filling the matrix, the algorithm performs a traceback to find the optimal alignment path. Starting from the bottom-right corner and moving towards the top-left corner, adjacent cells are examined in reverse order to determine the best path with the highest total score. The optimal alignment path is the one with the maximum score.
C. Word or k-tuple method
- Word or k-tuple methods are heuristic methods best known for their use in the database search tools FASTA and BLAST.
- The word method is a fast method for aligning two sequences. It begins by identifying short identical sequences, also known as words or k-tuples, and then uses dynamic programming to align the sequences based on these words.
Methods of Multiple Sequence Alignment
Multiple sequence alignment can be performed using either exhaustive or heuristic approaches.
A. Exhaustive algorithms
- Exhaustive alignment involves examining all possible alignments at once.
- A multidimensional search matrix is required to perform multiple sequence alignment using the exhaustive algorithm, similar to the two-dimensional matrix used in dynamic programming for pairwise alignment. This means that to align N sequences, an N-dimensional matrix is required.
- Dynamic programming is a powerful method for aligning sequences, but as the number of sequences to be aligned increases, the amount of computational time and memory space also increases. This means that the method becomes computationally impractical for large data sets. As a result, dynamic programming is typically only used for small data sets with fewer than ten short sequences.
- Heuristic approaches are typically used for larger data sets to achieve a more efficient alignment.
B. Heuristic algorithm
i. Progressive method
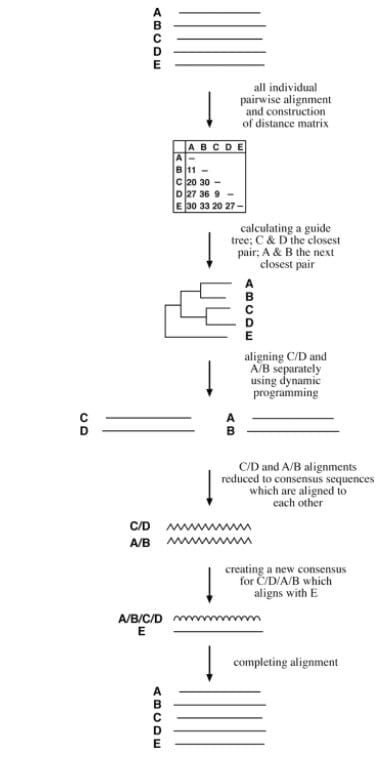
- The progressive method, also known as the tree-based algorithm, is a step-wise assembly of multiple alignments based on pairwise similarity. This method is called progressive because it aligns sequences in a step-wise manner.
- First, it performs pairwise alignments of all the sequences using the Needleman–Wunsch global alignment method and records the similarity scores.
- Then, it converts the scores into evolutionary distances to create a distance matrix. A guide tree is constructed from the distance matrix using the neighbor-joining method.
- The guide tree is used to direct the realignment of sequences based on their relative positions on the tree, starting with the two most closely related sequences and adding more distant sequences one at a time until all sequences are aligned.
- Clustal and T-Coffee are two well-known progressive alignment programs.
ii. Iterative Method
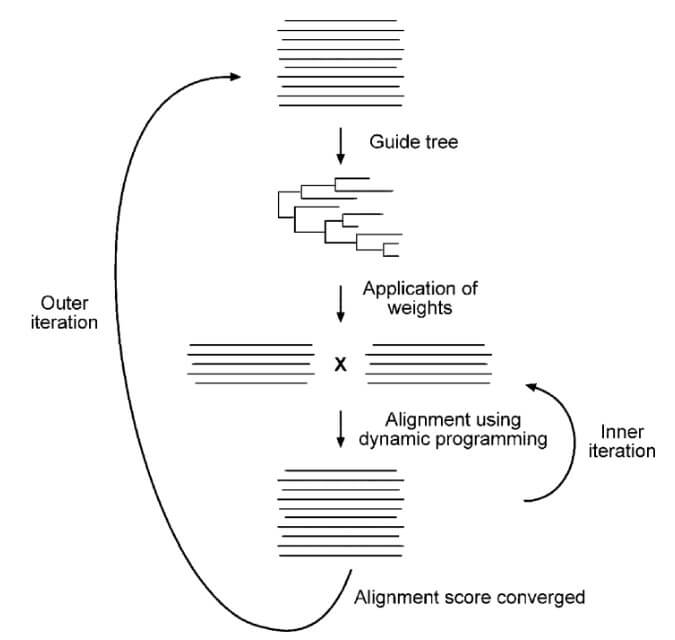
- The iterative method involves improving an initial suboptimal solution by repeatedly modifying it until an optimal solution is reached.
- An initial pairwise alignment is conducted to create a tree that provides weights for creating alignments. Aligned regions with gaps are identified and iteratively adjusted to enhance the alignment score. The highest-scoring alignment is used in a new set of calculations to predict a new tree, new weights, and new alignments. The procedure is repeated until there is no more improvement in the alignment score.
- PRRN is a web-based program that uses the iterative method of alignment.
iii. Block-based method
- The progressive and iterative alignment methods are based on global alignment and may not be effective in identifying conserved domains and motifs in highly divergent sequences of different lengths.
- To align such divergent sequences, a local alignment-based approach is needed.
- The block-based method is one such method that identifies a block of ungapped alignment that is shared by all sequences.
Applications of sequence alignment
- Sequence alignment can identify unknown sequences by comparing them with already known sequences in databases.
- Sequence alignment is also used to identify conserved sequence patterns and motifs, which helps to characterize the functions of the sequences.
- Sequence alignment can also produce phylogenetic trees and obtain information about the evolutionary relationship between the sequences aligned.
- Sequence alignment can also predict proteins’ secondary and tertiary structures. It can also predict gene locations and new members of gene families.
- Sequence alignment can also be used to develop degenerate PCR primers by analyzing multiple related sequences.
References
- Batzoglou, S. (2005). The many faces of sequence alignment. Briefings in Bioinformatics, 6(1), 6–22. doi:10.1093/bib/6.1.6
- https://www.ebi.ac.uk/Tools/psa/
- Mount, D. W. (2001) Bioinformatics: sequence and genome analysis. Cold Spring Harbor Laboratory Press.
- Naznin, F., Sarker, R., & Essam, D. (2012). Progressive Alignment Method Using Genetic Algorithm for Multiple Sequence Alignment. IEEE Transactions on Evolutionary Computation, 16(5), 615–631. doi:10.1109/tevc.2011.2162849
- Sequence Alignment – Handbook of Discrete and Combinatorial Mathematics – NCBI Bookshelf (nih.gov)
- Xiong, J. (2006). Essential Bioinformatics. Cambridge: Cambridge University Press. doi:10.1017/CBO9780511806087