With the increase in DNA and protein sequence databases, there is a growing need for more faster and efficient methods to analyze this large amount of data. One of the most commonly used bioinformatics tools today to study DNA and protein sequences is called BLAST.
BLAST stands for Basic Local Alignment Search Tool. It is a widely used bioinformatics program that was first introduced by Stephen Altschul et al. in 1990 and has since become one of the most popular tools for sequence similarity search.
BLAST is a powerful tool for analyzing biological sequence data. Since the initial release of BLAST in 1990, it has undergone continuous updates to improve its speed and accuracy. BLAST is now considered a crucial and widely used tool in the field of bioinformatics. It has played a vital role in numerous research studies and has paved the way for the development of other sequence comparison tools.
Interesting Science Videos
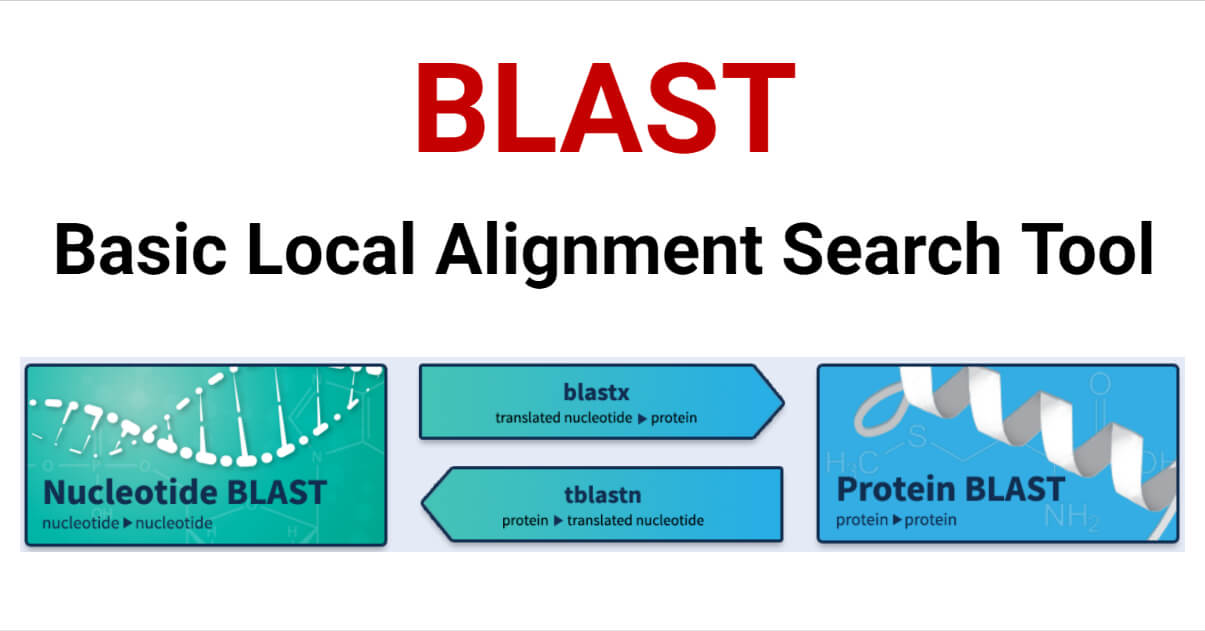
5 Types of BLAST
There are five types (variants) of BLAST that are differentiated based on the type of sequence (DNA or protein) of the query and database sequences.
- BLASTN compares a nucleotide query sequence to a nucleotide sequence database.
- BLASTP compares a protein query sequence to a protein sequence database.
- BLASTX compares a nucleotide query sequence to a protein sequence database by translating the query sequence into its six possible reading frames and aligning them with the protein sequences.
- TBLASTN compares a protein query sequence to a nucleotide sequence database by translating the nucleotide sequences in all six reading frames and aligning them with the protein sequence.
- TBLASTX compares a nucleotide query sequence to a nucleotide sequence database by translating the query sequence in all six reading frames and aligning them with the nucleotide sequences.
How BLAST Works
BLAST works by comparing a query sequence to a database of sequences to find regions of similarity. It uses a heuristic approach to search for similarities in the database, making it faster and more efficient.
BLAST performs sequence alignment through the following steps.
- Step 1: The first step is to create a lookup table or list of words from the query sequence. This step is also called seeding. First, BLAST takes the query sequence and breaks it into short segments called words. For protein sequences, each word is usually three amino acids long, and for DNA sequences, each word is usually eleven nucleotides long.

- Step 2: The second step is to search a database of known sequences to find any sequences that contain the same words as the query sequence. This is done to identify database sequences containing the matching words.

- Step 3: BLAST then scores the similarity of the matching words. The matching of the words is scored by a given substitution matrix. If a word is above a certain threshold, it is considered a match.
Two commonly used substitution matrices for protein sequences are PAM (Percent Accepted Mutations) and BLOSUM (Blocks Substitution Matrix). For nucleotide sequences, the scoring matrix is based on match-mismatch scoring. - Step 4: The fourth step involves pairwise alignment by extending the words in both directions while counting the alignment score using the same substitution matrix. If the score drops below a certain threshold due to differences in the sequences or mismatches, the alignment stops. The resulting aligned segment pair without gaps is called the high-scoring segment pair (HSP).
BLAST also calculates a statistical significance value for each alignment. It is called E-value or Expect value. The E-value represents the probability of obtaining a sequence match by random chance. A lower E-value indicates that the sequence match is less likely to be a result of random occurrence. Hence, the lower the E-value, the higher the level of significance.
Characteristics of BLAST
Several key features of BLAST make it a widely used tool in bioinformatics. Some of these are:
- BLAST is fast and efficient, making it possible to handle large databases of sequences.
- It is a flexible and versatile tool as it can be used to search for similarities in both nucleotide and protein sequences.
- It is highly sensitive which allows the identification of even small similarities between sequences.
- It aims to identify regions of local similarity between the query sequence and the database sequence, rather than attempting to align the entire sequences.
- It has a user-friendly interface that makes it easy to input query sequences and interpret the results.
Applications of BLAST
BLAST has a wide range of applications. Some of the most common applications are:
- BLAST can be used to identify unknown sequences by comparing them with known sequences in a database which helps in predicting the functions of proteins or genes.
- BLAST can also be used in phylogenetic analysis which is important for understanding the evolutionary relationships between different species.
- BLAST can also be used to identify functionally conserved domains within proteins which is important for predicting the functions of proteins.
References
- BLAST QuickStart – Comparative Genomics – NCBI Bookshelf (nih.gov)
- BLAST: Basic Local Alignment Search Tool (nih.gov)
- BLAST: Compare & identify sequences – NCBI Bioinformatics Resources: An Introduction – Library Guides at UC Berkeley
- Grzegorz M. Boratyn, Christiam Camacho, Peter S. Cooper, George Coulouris, Amelia Fong, Ning Ma, Thomas L. Madden, Wayne T. Matten, Scott D. McGinnis, Yuri Merezhuk, Yan Raytselis, Eric W. Sayers, Tao Tao, Jian Ye, Irena Zaretskaya, BLAST: a more efficient report with usability improvements, Nucleic Acids Research, Volume 41, Issue W1, 1 July 2013, Pages W29–W33, https://doi.org/10.1093/nar/gkt282
- How BLAST Works (nih.gov)
- McGinnis, S., & Madden, T. L. (2004). BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Research, 32(Web Server issue), W20. https://doi.org/10.1093/nar/gkh435
- NCBI_blast.pdf (unmc.edu)
- Xiong, J. (2006). Essential Bioinformatics. Cambridge: Cambridge University Press. doi:10.1017/CBO9780511806087