A phylogenetic tree (evolutionary tree) is the graphical representation of the evolutionary history of biological sequences and allows us to visualize the evolutionary relationships between them.
Evolution explains the diversity of life on Earth, and understanding evolutionary relationships helps us understand the origin and relationships between different organisms. Molecular phylogenetics is an important aspect of bioinformatics that helps us understand evolutionary history and relationships by using molecular data such as DNA or protein sequences to create phylogenetic trees.
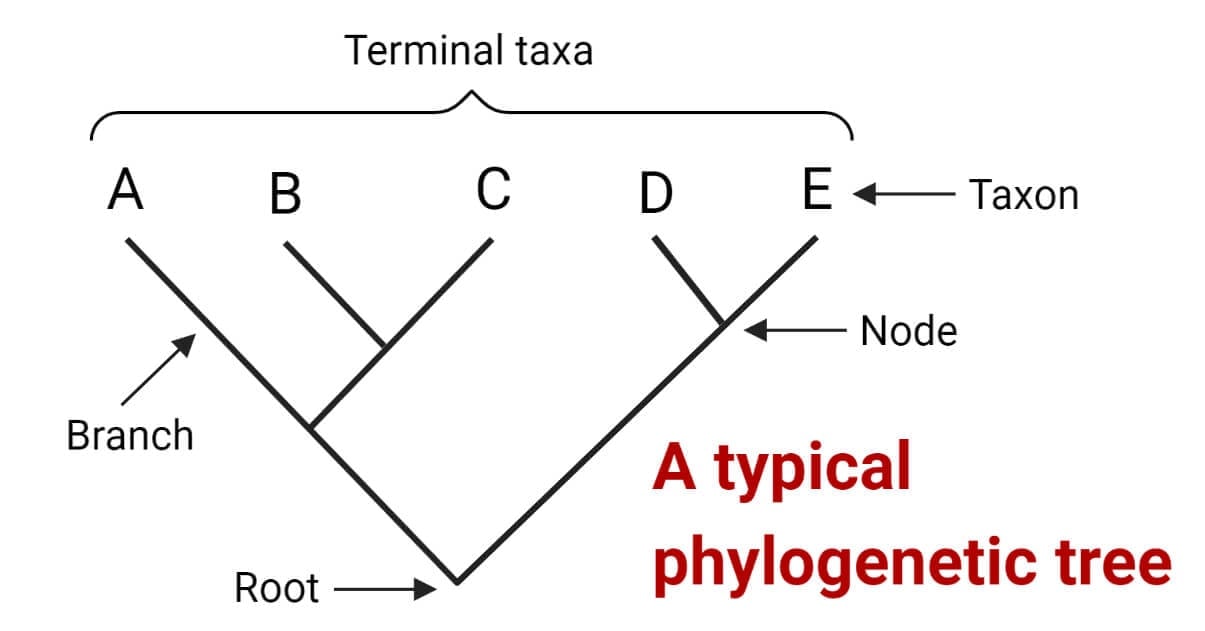
These trees are displayed as two-dimensional diagrams, with branching lines representing the evolutionary history and relatedness of different groups of organisms. The endpoints of the branches represent present-day species or sequences and are referred to as taxa or operational taxonomic units (OTUs). The connecting point where two branches come together is called a node, and it represents an inferred common ancestor of the organisms or sequences connected to the branches. The point where the tree divides into two parts at the bottom is known as the root node, and it represents the common ancestor of all the members of the tree.
Interesting Science Videos
Types of Phylogenetic Tree
There are several different types of phylogenetic trees. They can be classified as:
On the basis of the presence or absence of a common root
Rooted trees are trees that have a specified root node, which represents the common ancestor of all the organisms in the tree.
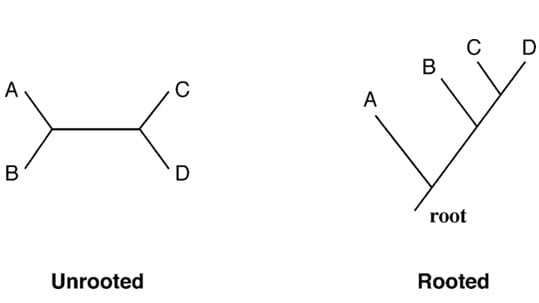
Unrooted trees do not have a specified root node and show only the branching pattern of the evolutionary relationships among taxa or OTUs, without any information about their common ancestor.
On the basis of topology
Cladogram is a type of phylogenetic tree that displays only the branching pattern of evolutionary relationships among organisms. Cladograms are unscaled, which means that the branch lengths do not reflect the amount of evolutionary divergence between taxa or operational taxonomic units (OTUs).
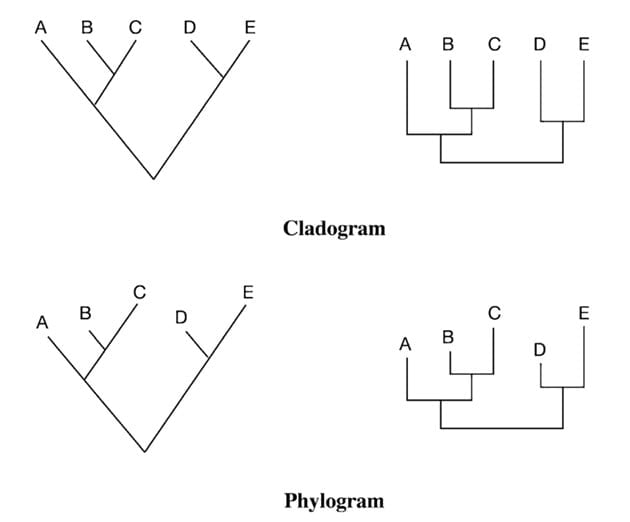
Phylogram is a type of phylogenetic tree that represents the evolutionary relationships among organisms by showing both the branching pattern and the amount of evolutionary divergence. Phylograms are scaled, which means that the branch lengths are proportional to the amount of evolutionary divergence.
Phylogenetic Tree Construction Steps
Phylogenetic tree construction is a complex process that involves several steps:
1. Selection of molecular marker
- The first step in constructing a phylogenetic tree is to choose the appropriate molecular marker.
- The choice of molecular marker depends on the characteristics of the sequences and the purpose of the study. Either nucleotide or protein sequence data can be used.
- For closely related organisms, nucleotide sequences are preferable, while for more divergent groups, slowly evolving nucleotide sequences or protein sequences may be used.
- Protein sequences are preferred over nucleotide sequences in many cases because they are more conserved and allow for more sensitive alignment due to having more characters.
- Although protein sequences offer several benefits for phylogenetic analysis, DNA sequences can also provide valuable information in certain instances, especially when dealing with closely related sequences.
2. Multiple sequence alignment
- After the selection of molecular markers, the next step is to align the sequences from different species.
- This is the most important step because the accuracy of the resulting phylogenetic tree depends on the quality of the alignment.
- Alignment programs such as T-Coffee can be used.
- Gblocks is one of the automatic programs that can help improve alignment by eliminating poorly aligned positions and divergent regions.
3. Selection of a model of evolution
- The third step of phylogenetic tree construction is the selection of an appropriate evolutionary model.
- Evolutionary (or substitution) models are statistical models that describe the substitution and divergence of sequences over time.
- There are several substitution models available for both nucleotide and amino acids.
- Two commonly used substitution models for nucleotides are the Jukes-Cantor (JC) model and Kimura’s two-parameter model.
- There are also many amino acid substitution models. The most commonly used ones are the Dayhoff model (PAM) and the Jones-Taylor-Thornton (JTT) model.
4. Construction of the phylogenetic tree
- The next step is the construction of the phylogenetic tree.
- The two main methods for constructing phylogenetic trees are distance-based and character-based methods.
- Distance-based methods rely on computing the amount of dissimilarity between sequences, while character-based methods use molecular sequences from individual taxa to trace the character states of the common ancestor.
5. Assessment of the reliability of the tree
- The final step involves assessing the reliability of the phylogenetic tree. This can be done by a statistical method called bootstrapping which is used to assess the reliability of a phylogenetic tree’s topology.
- It involves repeatedly resampling the initial sequence data to generate multiple subsets of derived sequences, referred to as bootstrap samples.
- These samples are then used to construct a new phylogenetic tree using the same method as the original tree.
- Interior branches that are accurately predicted by the new tree are assigned a value of 1. This process is repeated numerous times, and the percentage of times each interior branch receives a value of 1 is calculated as the bootstrap value or confidence value.
- A bootstrap value of 95 or more is generally considered to indicate an accurate topology, and these values are expressed as percentages on the branches of the phylogenetic tree.
- Besides bootstrapping, other resampling strategies like Jackknifing and Bayesian Simulation can also be used.
Read Also- How to construct a Phylogenetic tree?
Phylogenetic Tree Construction Methods
The methods to construct phylogenetic trees can be classified into two major types:
1. Distance-based methods
Distance-based tree construction methods involve calculating evolutionary distances between sequences by using substitution models, which are then used to construct a distance matrix. Using the distance matrix, a phylogenetic tree is constructed. The two popular distance-based methods are UPGMA and NJ.
a. Unweighted Pair Group Method with Arithmetic Mean (UPGMA)
- UPGMA is the simplest distance-based method that constructs a rooted phylogenetic tree using sequential clustering.
- First, all sequences are compared using pairwise alignment to calculate the distance matrix.
- Using this matrix, the two sequences with the smallest pairwise distance are clustered as a single pair. A node is placed at the midpoint between them.
- Next, the distance between this pair and all other sequences is recalculated to form a new matrix.
- This new matrix is used to identify and cluster the sequence that is closest to the first pair. This process is repeated until all sequences have been placed on the tree.
- UPGMA method assumes that the evolutionary rate of all taxa is constant, and they are equidistant from the root, indicating the presence of a molecular clock mechanism.
b. Neighbor-Joining (NJ)
- The neighbor-joining method is the most widely used distance-based method.
- It is similar to the UPGMA method in terms of building the tree using a distance matrix however, it does not assume the molecular clock and produces an unrooted tree.
- The neighbor-joining algorithm starts with a completely unresolved star tree, where all sequences are connected to a single node.
- It then iteratively adds branches between the two closest neighbors and the remaining sequences in the tree. The algorithm calculates the pairwise distances between all sequences and uses these distances to determine the closest neighbors.
- Once the closest neighbors are identified, the algorithm consolidates them into a new node, effectively reforming the star tree. This process is repeated until all sequences are connected in a fully resolved tree.
2. Character-Based Methods
Character-based methods involve analyzing sequence data by directly examining the sequence characters, rather than relying on pairwise distance comparisons. These methods evaluate all sequences at once by analyzing one character or site at a time.
Character-based methods are generally considered more accurate than distance-based methods. However, character-based methods are more computationally intensive and require more sophisticated statistical models.
The maximum parsimony (MP) and maximum likelihood (ML) methods are the two most commonly used character-based tree construction methods.
a. Maximum parsimony (MP)
- Maximum parsimony method is a character-based method that selects the tree with the least number of evolutionary changes or the shortest total branch length.
- Initially, multiple sequence alignment is performed to identify potential positions in the sequences that correspond to each other.
- Each aligned position is analyzed to identify the trees that require the smallest number of evolutionary changes to produce the observed sequence changes.
- This process is repeated for all positions in the sequence alignment, and the trees that produce the lowest overall number of changes for all positions are selected.
- This method works best for relatively similar sequences and for small numbers of sequences.
b. Maximum likelihood (ML)
- Maximum likelihood is a statistical method that uses probabilistic models to identify the most appropriate tree that has the maximum probability of generating the observed data.
- Similar to the maximum parsimony method, this approach evaluates each column of a multiple sequence alignment during the analysis.
- However, unlike maximum parsimony, ML considers all possible trees that could explain the observed data.
- The likelihood of each possible tree is calculated, and the tree with the highest probability is selected as the most likely evolutionary history of the sequences.
Applications of the phylogenetic tree
Phylogenetic trees have various practical applications, including:
- Phylogenetic trees can be used to study the evolutionary relationships between different species and to understand the evolutionary processes over time.
- Phylogenetic trees can be used to study the diversity and distribution of species and to develop conservation strategies to protect endangered species and ecosystems.
- Phylogenetic trees can be used to identify the origins of pathogens and to track the spread of diseases.
- Phylogenetic trees can also be used in forensics to identify the origins of biological samples found at crime scenes and to link suspects to crimes.
- Phylogenetic trees are useful for organizing and classifying organisms and species according to their DNA sequences and morphological similarities and differences.
References
- Bawono, P., & Heringa, J. (2014). Phylogenetic Analyses. Comprehensive Biomedical Physics, 93–110. doi:10.1016/b978-0-444-53632-7.01108-4
- Choudhuri, S. (2014). Phylogenetic Analysis. Bioinformatics for Beginners, 209–218. doi:10.1016/b978-0-12-410471-6.00009-8
- https://www.ebi.ac.uk/training/online/courses/introduction-to-phylogenetics/why-is-phylogenetics-important/
- https://www.geeksforgeeks.org/phylogenetic-tree/
- https://www.nature.com/scitable/topicpage/reading-a-phylogenetic-tree-the-meaning-of-41956/
- Mount, D. W. (2001) Bioinformatics: sequence and genome analysis. Cold Spring Harbor Laboratory Press.
- Munjal, G., Hanmandlu, M., & Srivastava, S. (2019). Phylogenetics Algorithms and Applications. Ambient Communications and Computer Systems, 904, 187-194. https://doi.org/10.1007/978-981-13-5934-7_17
- Scott, A. D., & Baum, D. A. (2016). Phylogenetic Tree. Encyclopedia of Evolutionary Biology, 270–276. doi:10.1016/b978-0-12-800049-6.00203-1
- Xiong, J. (2006). Essential Bioinformatics. Cambridge: Cambridge University Press. doi:10.1017/CBO9780511806087