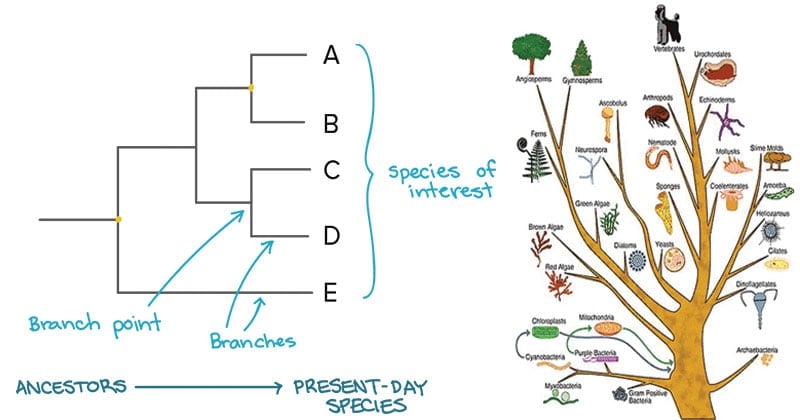
- A phylogenetic tree is a visual representation of the relationship between different organisms, showing the path through evolutionary time from a common ancestor to different descendants.
- Similarities and divergence among related biological sequences revealed by sequence alignment often have to be rationalized and visualized in the context of phylogenetic trees. Thus, molecular phylogenetics is a fundamental aspect of bioinformatics.
- Molecular phylogenetics is the branch of phylogeny that analyzes genetic, hereditary molecular differences, predominately in DNA sequences, to gain information on an organism’s evolutionary relationships.
- The similarity of biological functions and molecular mechanisms in living organisms strongly suggests that species descended from a common ancestor. Molecular phylogenetics uses the structure and function of molecules and how they change over time to infer these evolutionary relationships.
- From these analyses, it is possible to determine the processes by which diversity among species has been achieved. The result of a molecular phylogenetic analysis is expressed in a phylogenetic tree.
Phylogenetic Analysis and the Role of Bioinformatics
Molecular data that are in the form of DNA or protein sequences can also provide very useful evolutionary perspectives of existing organisms because, as organisms evolve, the genetic materials accumulate mutations over time causing phenotypic changes. Because genes are the medium for recording the accumulated mutations, they can serve as molecular fossils. Through comparative analysis of the molecular fossils from a number of related organisms, the evolutionary history of the genes and even the organisms can be revealed.
However, phylogeny inference are notoriously difficult endeavours because the number of solutions increases explosively with the number of taxa and the tremendous number of new questions in evolutionary biology that could be investigated through the use of larger taxon samplings.
But with the development and use of computational and an array of bioinformatics tools, the ability to analyze large data sets in practical computing times, and yielding an optimal or near-optimal solutions with high probability are being possible. In response to this trend, much of the current research in phyloinformatics (i.e., computational phylogenetics) concentrates on the development of more efficient heuristic approaches.
Steps in Phylogenetic Analysis

The basic steps in any phylogenetic analysis include:
- Assemble and align a dataset
- The first step is to identify a protein or DNA sequence of interest and assemble a dataset consisting of other related sequences.
- DNA sequences of interest can be retrieved using NCBI BLAST or similar search tools.
- Once sequences are selected and retrieved, multiple sequence alignment is created.
- This involves arranging a set of sequences in a matrix to identify regions of homology.
- There are many websites and software programs, such as ClustalW, MSA, MAFFT, and T-Coffee, designed to perform multiple sequence on a given set of molecular data.
- Build (estimate) phylogenetic trees from sequences using computational methods and stochastic models
- To build phylogenetic trees, statistical methods are applied to determine the tree topology and calculate the branch lengths that best describe the phylogenetic relationships of the aligned sequences in a dataset.
- The most common computational methods applied include distance-matrix methods, and discrete data methods, such as maximum parsimony and maximum likelihood.
- There are several software packages, such as Paup, PAML, PHYLIP, that apply these most popular methods.
- Statistically test and assess the estimated trees.
- Tree estimating algorithms generate one or more optimal trees.
- This set of possible trees is subjected to a series of statistical tests to evaluate whether one tree is better than another – and if the proposed phylogeny is reasonable.
- Common methods for assessing trees include the Bootstrap and Jackknife Resampling methods, and analytical methods, such as parsimony, distance, and likelihood.
Bioinformatics Tools for Phylogenetic Analysis
- There are several bioinformatics tools and databases that can be used for phylogenetic analysis.
- These include PANTHER, P-Pod, PFam, TreeFam, and the PhyloFacts structural phylogenomic encyclopedia.
- Each of these databases uses different algorithms and draws on different sources for sequence information, and therefore the trees estimated by PANTHER, for example, may differ significantly from those generated by P-Pod or PFam.
- As with all bioinformatics tools of this type, it is important to test different methods, compare the results, then determine which database works best (according to consensus results) for studies involving different types of datasets.
References
- Xiong J. (2006). Essential Bioinformatics. Texas A & M University. Cambridge University Press.
- Arthur M Lesk (2014). Introduction to bioinformatics. Oxford University Press. Oxford, United Kingdom.
- Brown, D, K Sjölander (2006) “Functional Classification Using Phylogenomic Inference.” PLos Computational Biology, 2(6):0479-0483.
- http://www.math.umaine.edu/~khalil/courses/MAT500/papers/MAT500_Paper_Phylogenetics.pdf
- https://www.ncbi.nlm.nih.gov/books/NBK21122/’
- http://www.bioinfbook.org/php/?q=chapter7
- http://previouslife.lanevol.org/LANE/Molecular_Phylogenetics.html
- https://www.slideshare.net/AjayChandra17/molecular-phylogenetics