Protein structure prediction is an important aspect of computational biology that aims to determine the three-dimensional (3D) structure of a protein. Homology modeling is one of the computational methods of protein structure prediction.
Homology modeling, also known as comparative modeling, is a method used to predict the 3D structure of a protein with an unknown structure by using the known structure of a homologous protein.
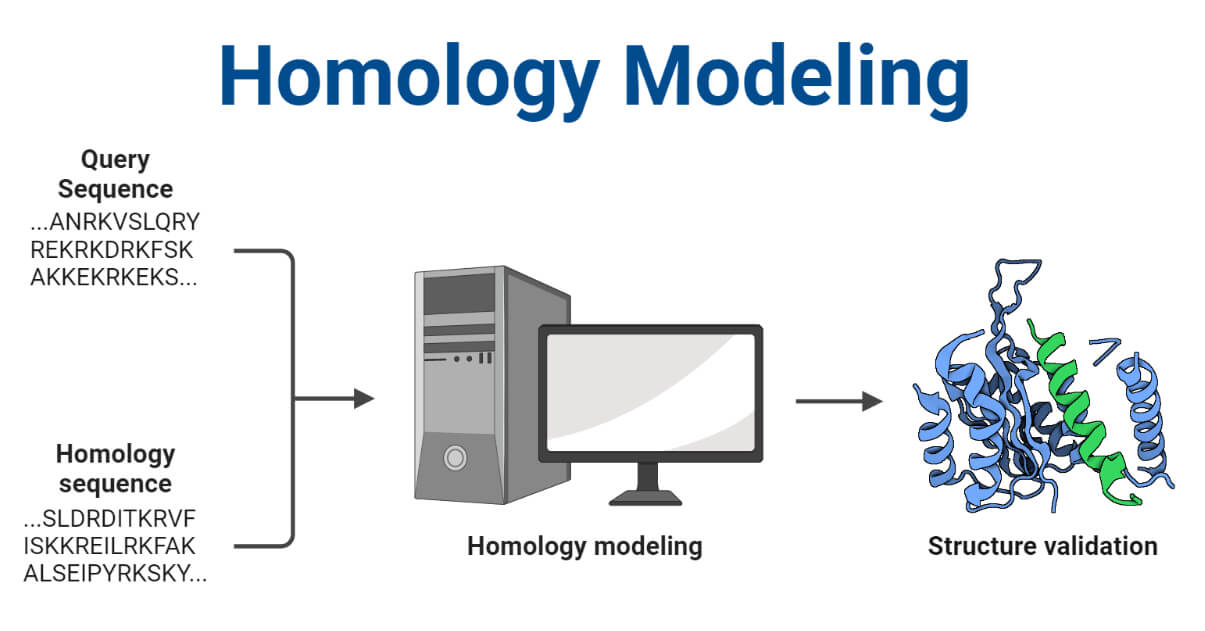
The method relies on the fact that the 3D structure of proteins is often better conserved than their amino acid sequence. Therefore, proteins with similar sequences are likely to have similar structures.
This method is becoming increasingly important as the number of experimentally determined structures is limited compared to the number of protein sequences available. As the gap between the number of protein sequences and experimentally determined structures continues to grow, homology modeling has an increasingly important role to play in filling this gap.
Interesting Science Videos
Steps in Homology Modeling
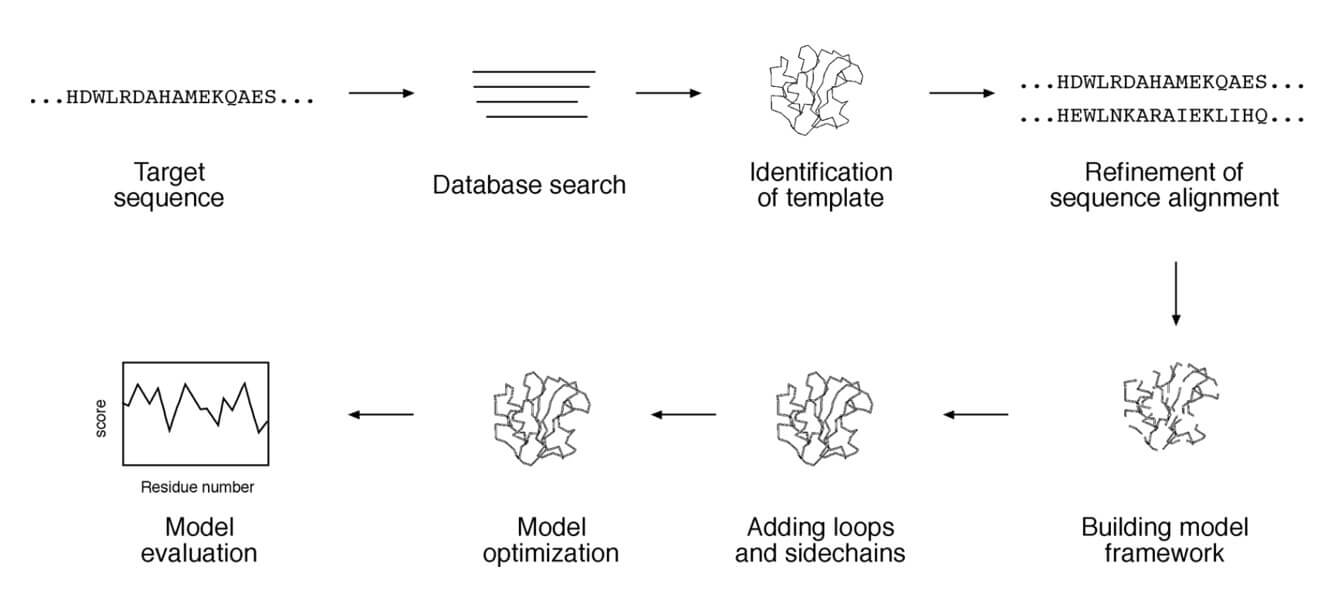
The overall homology modeling procedure consists of a series of steps:
- Identification and Selection of Template Structures:
- The first step in this process is identifying and selecting appropriate template structures from the Protein Data Bank (PDB).
- To identify appropriate templates, pair-wise sequence alignment methods such as BLAST can be used to identify proteins with high sequence homology to the query protein in the PDB.
- More sensitive methods involve performing multiple sequence alignments or using automated iterative methods such as PSI-BLAST.
- If the homology between the protein of interest and the potential templates is low (below 35%), alternative methods such as profile-profile alignments, and Hidden Markov Models (HMMs) are used to reduce gaps in the alignment.
- Once several templates are identified, the most appropriate ones for modeling must be chosen. Factors such as sequence identity and similarity, alignment scores, and phylogenetic relationships should be considered, as well as other information such as biological function and environmental context.
- Alignment of the Query Sequence with the Template:
- In homology modeling, the alignment of the query sequence with the template is an important step in the process.
- It is essential to achieve a high-quality sequence alignment as it directly impacts the accuracy of the resulting model. A homology model can only be as good as the sequence alignment.
- Multiple sequence alignment is preferred as it contains evolutionary information that is important for structure and function. Effective multiple sequence alignment programs like Proline and T-Coffee are recommended for use.
- However, even the best program may contain errors, so it is necessary to visually examine the alignment to confirm that conserved residues are aligned correctly.
- In case of inaccuracies, manual adjustments may be necessary, including repositioning gaps, changing substitution scores, or moving sequence segments to improve alignment quality.
- Building a Three-Dimensional Model of the Query Protein:
- Sequence alignment is followed by the 3D model building of the target proteins.
- There are different methods used for generating 3D models of protein based on its templates. These methods are classified into four categories: rigid-body assembly, segment matching, spatial restraint, and artificial evolution methods.
- In rigid-body assembly, the protein structure is broken down into basic conserved core regions, loops, and side chains. The rigid body parts are picked up from the template protein structures and brought together using tools like 3D-JIGSAW, BUILDER, and SWISS-MODEL.
- Segment matching involves using a cluster of atomic positions from template structures as leading positions. These atomic positions are used to select segments from known structures in the database based on sequence identity, geometry, and energy. These segments are aligned with the template structure to create a model of the target protein. This can be done with SEGMOD/ENCAD.
- The spatial restraint method builds protein structures by applying spatial restraints to guide the building of a model that closely matches the template structure. The restraints are determined by stereochemical principles on bond length, bond angle, dihedral angles, and van der Waals contact distances. MODELLER is a software package commonly used to perform this method.
- The artificial evolution method involves using the rigid-body assembly method and stepwise template evolutionary mutations together until the template sequence is the same as the target sequence. This can be performed with NEST.
- Loop Modeling
- There are often gaps or insertions that can occur in sequence alignments when modeling proteins. These gaps are called loops and are structurally less conserved during evolution.
- Loop modeling is an important step in protein structure prediction because loops play a crucial role in determining the function of a protein. However, predicting their structure is a complex process.
- There are two main methods for loop prediction: the database search method and the conformational search method.
- The database search method involves comparing the target protein sequence with all known protein structures to identify segments that can be used to model the loop.
- The conformational search approach involves optimizing a scoring function to find the best possible loop conformation. This method generates many random loops and searches for the one that fits the target protein structure. This approach is also called ab initio because it does not rely on pre-existing structural information.
- There are specialized programs available for loop modeling, such as FREAD, PETRA, and CODA.
- Side Chain Modeling
- Side chain modeling is the process of predicting the conformation of the side chains of amino acids in a protein structure. This process is crucial in evaluating protein-ligand and protein-protein interactions.
- This is usually done by placing the side chains onto the backbone coordinates that are derived from a parent structure or from ab initio modeling simulations.
- Searching every possible conformation of a side chain is computationally time-consuming and not effective so, most side chain prediction programs use the preferred conformation called rotamers. These rotamers are stored in a rotamer library, which is a collection of preferred side chain conformations ranked by their frequency of occurrence.
- Different energy functions and search strategies are used to select the most appropriate rotamer for each amino acid side chain based on the preferred protein sequence and the given backbone coordinates.
- There are various tools available for side chain modeling, such as RAMP and SCWRL.
- Model optimization
- The next step is to optimize the model. Optimization of a model refers to the process of refining the initial model to improve its accuracy and reliability.
- This optimization process involves adjusting the positions of the atoms in the model to reduce any clashes or steric hindrances between atoms.
- Energy minimization process is one of the methods of model optimization. It calculates the potential energy of the model and adjusts the positions of the atoms to minimize this energy. Excessive energy minimization can often move residues away from their correct positions, so only limited energy minimization is recommended.
- In addition to energy minimization, other optimization techniques such as molecular dynamics and Monte Carlo simulations can be used.
- Molecular dynamics simulations involve simulating the movements and interactions of the atoms in the model over time, which allows the exploration of different conformations and the identification of the most stable or energetically favorable ones.
- Monte Carlo simulations use random sampling to explore different configurations of the molecule and identify the most stable or energetically favorable ones.
- Model Validation and Evaluation
- After the 3D model of the query protein has been built, it is essential to validate and evaluate its quality to ensure that it is biologically relevant and can be used for further studies.
- This involves comparing the model to real experimental 3D structures and using various evaluation methods to assess the model’s stereochemistry, physical parameters, knowledge-based parameters, statistical mechanics, and other criteria.
- Some commonly used programs for evaluating protein structures are WHATIF, PROCHECK, and PROSA. These programs compare the properties of the homology model to those found in the experimental structures in the PDB.
- The quality of the homology model largely depends on the accuracy of the sequence alignment and the parts of the protein where the template is not similar to the query protein.
Applications of Homology Modeling
Homology modeling has many practical applications:
- Homology modeling is important for the prediction of protein structures that are difficult to obtain experimentally using techniques like X-ray crystallography or NMR.
- Homology modeling is used to predict the functional properties of proteins with unknown structures by modeling the protein structure and comparing it with known structures of related proteins.
- Homology modeling is also useful in studying the evolution of protein families by comparing the structures of related proteins from different species to identify the changes that have occurred during evolution.
- Homology modeling can be used to identify potential drug targets by predicting the structure of a protein to understand its function and identify potential sites for drug binding. This information can be used to design new drugs or optimize existing ones.
References
- Hameduh, T., Haddad, Y., Adam, V., & Heger, Z. (2020). Homology modeling in the time of collective and artificial intelligence. Computational and Structural Biotechnology Journal, 18, 3494-3506. https://doi.org/10.1016/j.csbj.2020.11.007
- Muhammed, M. T., & Aki-Yalcin, E. (2019). Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chemical biology & drug design, 93(1), 12–20. https://doi.org/10.1111/cbdd.13388
- Nilges, M. (2005). Homology Modeling. In: Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine. Springer, Berlin, Heidelberg. https://doi.org/10.1007/3-540-29623-9_5500
- Vyas, V. K., Ukawala, R. D., Ghate, M., & Chintha, C. (2012). Homology Modeling a Fast Tool for Drug Discovery: Current Perspectives. Indian Journal of Pharmaceutical Sciences, 74(1), 1-17. https://doi.org/10.4103/0250-474X.102537
- Xiong, J. (2006). Essential Bioinformatics. Cambridge: Cambridge University Press. doi:10.1017/CBO9780511806087