The concept of CADD attracts attention due to its higher selectivity, efficiency, efficacy, short time duration, low toxicity, and better to fit with various pharmacokinetic parameters (Makrynitsa et al., 2018) The proper balance of pharmaceutical chemistry with the biological activity is found to be the most critical for effective drug designing. With the ever-increasing demand for drugs, the selection of biologically active structures draws greater attention. The knowledge of structural biology and the power of computers has become possible to exploit the use of computational methods to identify active libraries to resolve the molecular obesity problem.
Interesting Science Videos
What is Computer-aided drug design (CADD)?
The primary objective of CADD is to screen, optimize and evaluate the activity of the compound against the target. It forms the multi-disciplinary approach for both academic and major pharmaceutical companies for better efficacy with no/fewer side effects. The advancement in CADD is based on similarity, target identification, structure prediction, binding site/ cavity, validation, understanding the protein-ligand interaction along with screening vast set of compounds, grasping the molecular dynamics simulations based on physiological conditions, Tallying with ADMET properties, estimating biological activity using QSAR (Qualitative structure-based assessment relationship) which is required for the lead compound for better efficacy and selectivity.
List of the drugs based on the computer-aided drug designing
| Drug | Disease | Target |
| Oxymorphone | Opioid analgesic | Agonist of mu- opiniod receptor |
| Saquinavir | AIDS | Inhibits proteases of HIV1 and HIV 2 |
| Captopril | Hypertension or high BP | Inhibits conversion of angiotensin-converting enzyme |
| Zanamivir | Affects influenza A and influenza B | Inhibits neuraminidase |
| Dorzolamide | Glaucoma and ocular hypertension | Inhibits carbonic anhydrase |
Types of CADD
A. Structure-Based Drug Designing (SBDD)
What is Structure-based Drug Designing (SBDD)?
The most efficient and powerful process which governs entire drug discovery is structure-based drug designing (SBDD). The information about the target of small molecules, genetic information with their sequences, binding information, cytotoxicity, absorption, metabolism, excretion(ADMET) data, and other important biological information serve as the most efficient sources for accelerating the drug discovery process. This has become a promising computational technique used by varied R&D, pharmaceutical companies. The pipeline involves the prediction of structure, visualization, binding site characterization, molecular docking and virtual screening, visualization of docked complex structure and their stability analysis, ADMET screening, and binding-free energy- MM PBSA will be involved.
Steps or Process of Structure-Based CADD
- Determination of protein structure
- Comparative modeling
- Identification of binding pocket
- Scoring function
- Knowledge-based scoring
- Force-based scoring function
- Protein-ligand docking algorithms
- Virtual screening
- Visualization of the protein-ligand diagram.
Softwares for Structure-Based Drug Designing (SBDD)
List of tools and applications in the SBDD approach
| Stages | Tools used | Brief Description | Links |
| 1. Target modeling | SWISS-MODEL | Homology modeling | SWISS-MODEL (expasy.org) |
| MODELER | Homology Modelling | MODELLER (salilab.org) | |
| Phyre and Phyre2 | Template detection alignment as well as 3D modeling | PHYRE2 Protein Fold Recognition Server (ic. ac. the UK) | |
| HHpred | Template detection, alignment, and 3D modeling | The HHpred interactive server for protein homology detection and structure prediction | HSLS (pitt.edu) | |
| I-TASSER | Threading ab initio modeling | I-TASSER server for protein structure and function prediction (zhanggroup.org) | |
| 2. Binding site | CASTp | Binding site prediction | CASTp 3.0: Computed Atlas of Surface Topography of proteins (uic.edu) |
| Active site prediction tool | Active site prediction | ACTIVE SITE PREDICTION SERVER (scfbio-iitd.res.in) | |
| 3. Molecular Docking | AutoDock Vina | Molecular docking and virtual screening | AutoDock Vina (scripps.edu) |
| Schrodinger | Maestro | Maestro | Schrödinger (schrodinger.com) | |
| 4.ADMET Prediction | admetSAR | ADMET prediction | Computational tools for ADMET (osdd.net) |
| QIKPROP | ADME analysis | QikProp | Schrödinger (schrodinger.com) | |
5. Molecular Dynamics Simulation | AMBER | Performs biomolecular simulation programs | www.gromacs.org |
| GROMACS | General Purpose molecular dynamic simulation program | https://www.gromacs.org | |
MM-PBSA | g_mmpbsa | This tool calculates binding energy using the MM-PBSA method | ambermd.org |
The binding site and prediction of cavity:
The most reliable and accurate region in the 3D structure in the protein-ligand complex is the analysis and prediction of the region in a protein where ligand can occupy its best pose. Several cavity prediction tools, such as Castro, Q-site Finder, and COACH, can provide the probable location of the binding site in a protein. These tools were designed based on evolutionary, energy-based, or geometric-based approaches. The evolutionary-based approach takes the information on all related proteins and shows nearly the same binding site. Geometry-based approaches are based on the features, such as shape, hydrophobic surface, and charged surface residues, whereas an energy-based approach uses a probe to identify the favorable binding regions on a protein.
Examples of Structure-Based Drug Designing (SBDD)
Listing important database resources in SBDD
| Database | Description | Link |
| Chemspider | Ligand structure | ChemSpider | Search and share chemistry |
| PubChem | Database with small molecules along with biological activity | PubChem (nih.gov) |
| DrugBank | FDA approved drugs for repurposing | DrugBank Online | Database for Drug and Drug Target Info |
| ZINC | The database provides compounds for virtual screening and molecular docking | ZINC (docking.org) |
Future Perspective
The combination of Artificial Intelligence and Machine Learning with structure-based techniques emerged as a new tool with immense potential in the discovery of therapeutics. Perhaps, the scientific community has to overcome the possibility associated with the algorithms, scoring functions, and the availability of existing data. This fundamental platform forms accelerate clinical trials and limit the failure of promising drug candidates. Later on, SBDD can emerge as a novel, reliable, and efficient technique for drug designing.
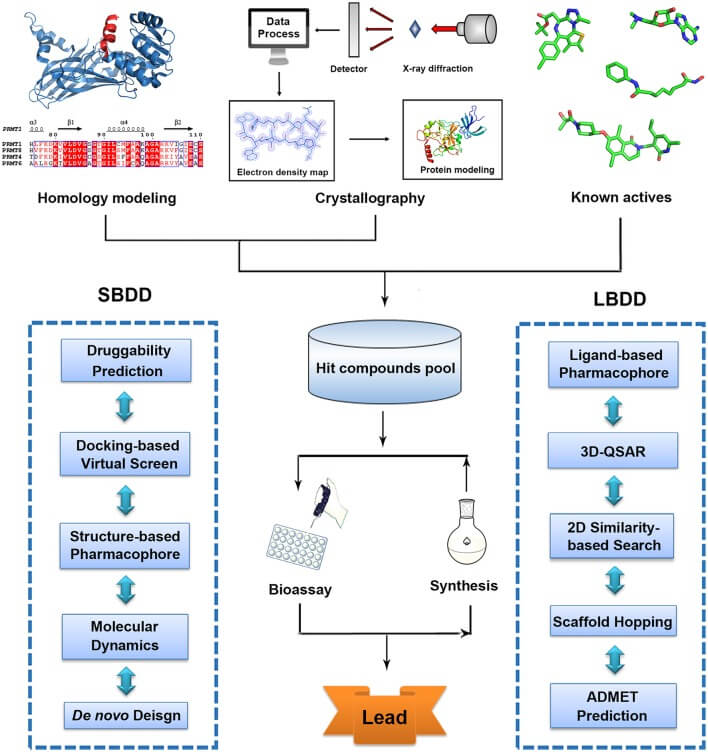
B. Ligand-based drug designing (LBDD)
What is Ligand-based drug designing (LBDD)?
The absence of a 3D macromolecular structure leads to very systematic target recognition using advanced computational tools. The present pharmaceutical researchers have faced a tougher situation and major challenge in this regard. The hypothesis of high-throughput screening(HTS) has been strengthened with better emerging strategies. The indirect mode of drug designing or ligand-based drug designing has supported widely this tougher challenge. This can be approached in three different ways such as
- Machine learning approach
- Similarity Search methods
- Pharmacophore models mapping
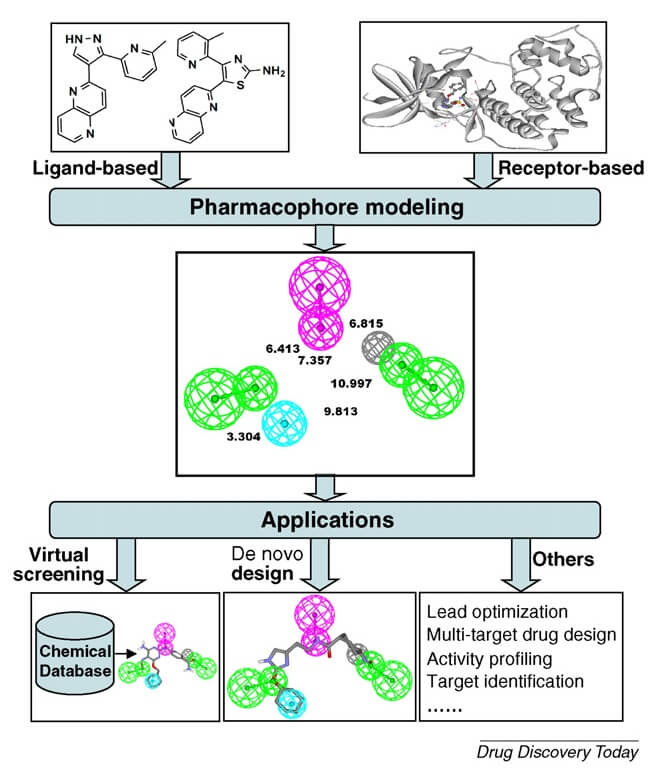
Pharmacophore models mapping
This approach is used in lead optimization in medical and computational chemistry. It is built upon using structural alignment and prediction activity and 3D database searching. The amount of known/unknown data set used for developing the molecular structure along with its activity towards any target. This can be implied in fields like ADMET profiling, estimation of side effects, off-target prediction. Taking the knowledge of pharmacophore, simulation studies were been improved by virtual screening.
Fundamental steps used in pharmacophore modeling
- Diverse set of structures with properly defined experimental activity
- Generation of conformations
- Employing 3D pharmacophore models to a training set of compounds in three phases
- Constructive Phase: Pharmacophore models were built on common active molecules
- Phase of Subtraction: Subtracts unlike models of pharmacophore
- Phase of optimization: Generate the hypothesis by optimizing a model
- Quality assessment: The selection of pharmacophore is laid on basis of the cost function
- Validation
- Fisher’s randomization test is performed for a validated set of compounds
- Finally, mapping is used for the test set of compounds.
Tools used for Pharmacophore modeling and applications
S.No | Tools | Description | Links |
1. | Pharmer | This tool uses compounds for virtual screening | Welcome to ZINCPharmer (pitt.edu) |
2. | Pharmapper | It implies knowledge of more than 7000 target-based pharmacophore models and provides the best match for input as a ligand against pharmacophore-based models. | Welcome to our lab (lab-request.cn) |
3. | pharmacist | This tool searches from a set of ligands in the absence of the target | BioInfo3D Group (tau.ac.il) |
4. | Pharmit tool | This relies on drug monitoring on Pharmacokinetics | pharmit: interactive exploration of chemical space (pitt.edu) |
5. | Discovery Studio | A tool used for therapeutics and drug monitoring | Free Download: BIOVIA Discovery Studio Visualizer – Dassault Systèmes (3ds.com) |
6. | Ligandscout | It models 3D pharmacophores from structural data either as training/test set of molecules | Linux |
7. | Phase | This tool considers both ligand and structure-based drug designing | Schrödinger | Schrödinger is the scientific leader in developing state-of-the-art chemical simulation software for use in pharmaceutical, biotechnology, and materials research. (schrodinger.com) |
Discovery of the lead molecule
The chemical compound which exhibits biological or pharmacological properties with therapeutic characteristics can be called a lead molecule. Parameters like good selectivity, efficiency, better efficacy along with pharma kinetic tools. Nevertheless, most of the lead compounds undergo structural modification to meet their certain biological properties, so-called lead optimization. However, the investigated lead compound has to satisfy five characteristics to act as a bioactive drug molecule. They are potency, duration, safety, availability, and pharmaceutical acceptability, where potency involves the capability of any molecule to exhibit desirable pharmaceutical properties in smaller quantities. Bioavailability marks the transportation of drug compounds with multiple barriers to reach the target site is called bioavailability. The concept of duration marks the time entry of drug molecules along with their pharmacological response. The safety rules involve the toxicity parameters which show less or no side effects on the target organisms. Henceforth all the above-mentioned factors contribute equally to the lead optimization of the drug molecule. The aim of rapid discovery of drugs involves target identification and validation. Upon target validation process, identification of hits and lead discovery phases has to be developed for a novel drug discovery process. Although, physical and biochemical parameters were used to decide the change in the structural property of the compounds to synthesize an effective lead molecule for the development of the drug. Several natural leads have become available in various databases and the literature with biological activity against its specific target, which leads to chemical modification. They fall into 3 categories such as natural, synthetic, and semi-synthetic.
Lead optimization and its strategies
Any lead compound can be modified structurally and maintain its physiochemical properties, thermodynamic, pharmacokinetic parameters, and toxicity (Jogensen, 2009). As per Lipinski’s rule of fie, the drug has to obey a few characteristics such as less than 5 hydrogen bonds and 10 acceptor bonds. Molecular mass should be less than 500Da and its partition coefficient( p- log) shouldn’t be higher than 5 (Lipinsk et al., 2003). The optimization of leads depends only on binding parameters which exhibit information about both drug efficacy and also on the metabolic process that occurs in living organisms. The branch of organic synthetic chemistry involves simplification of the structure, modification, dynamic and kinetics parameters, functional group interconversion, and bonding selectivity/ strength. Other parameters like kinetics include thermodynamics, pharmacodynamics along with higher improvement in ADMET properties have also to be considered.
Below is the list of natural product active compounds and their applications
| S.N. | Natural compounds | Active analogs | Application |
| 1. | Morpholine | Butorphanol, pentazocine, methadone | Targets opioid receptor agonist and antagonist |
| 2. | Halichondrin B | Eribulin, mesylate | Antitumor activity |
| 3. | Myriocin | Fingolimod | Immunosuppressive agent |
| 4. | Trichostatin A | Vorinstat | Histone deacetylase inhibitor |
| 5. | Schisandin C | Bicyclol | Anti hepatitis B activity |
| 6. | Asperlicin | Devazepide | Cholecystokinin receptor antagonist. |
Recent trends in drug designing
The repurposing of drugs ( drug repositioning, reprofiling, or retasking) is a way of approach to identify all the potential applications which are beyond the actual medical indication or investigational drugs(Ashburn & Thor, 2004). The concept can be referred to as drug reprofiling as uncovering new indications of the either of approved/ failed/ abandoned compounds to put in use for different diseases.
Conclusion
CADD functions as a multidisciplinary approach for the development of drugs in modern times. One of the major challenges is to develop a drug that well suits patients with better efficacy. CADD approaches have become more precise and efficient. A significant improvement in compound searching with the platform of similarity search, identification of the target, prediction of the structure, binding site activity prediction/validation, knowledge of protein-protein interaction under physiological conditions, calculating ADMET properties, biological activity estimation using tools like QSAR, above all guiding the necessary changes for a lead to improve efficacy as well selectivity. The existing modern drug discovery implies the knowledge of genomics, bioinformatics, computational chemistry, and combinatorial chemistry for virtual screening, HTS, Lenovo ligands. However, the concept of LBDD relies mostly on drug planning, which develops a simple drug molecule, a 3D through trial and error methods that improves the overall biological activity of the compound. This article has addressed a few software and fundamentals of ligand-based drug designing.
Applications of Computer-aided drug design (CADD)
The whole background in CADD draws much attention relaying on three major factors 1) screening of large set of molecules on target structure, which is evaluated by computationally as well as experimentally, 2) guiding the optimization of lead compounds depending on their affinity, pharmacokinetic parameters, as well toxicity 3) designing the novel compounds depending on their structure to improve their functionality as a drug compound. The applicability of CADD for modeling of the drug considers combinatorial chemistry and bioinformatics which address the major issues including cost and time duration.
Limitations of Computer-aided drug design (CADD)
Although the SBDD approach has proved as a pioneer in drug designing, it has to cross the challenges that the community has to look upon. This enhancement includes screening methods, chemogenomic compounds, data improvement, quantity, quality of various tools and databases, modifying in multitarget drug structures, toxicity predicting algorithms, integrating the approach for better efficacy and compatibility. The other most sounding parameter is electrostatic interactions, entropy calculations were ignored completely. Above all, there is no single software or package that works well for particular targets and ligands including the water molecule, and probable confirmation for the target and other innovations are still need to be addressed.
References
- Suchitra M. Ajjarapu, Apoorv Tiwari, Pramod Wasudeo Ramteke, Dev Bukhsh Singh, Sundip Kumar, Chapter 15 – Ligand-based drug designing, Editor(s): Dev Bukhsh Singh, Rajesh Kumar Pathak, Bioinformatics, Academic Press,2022, Pages 233-252.
- Yang SY. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today. 2010 Jun;15(11-12):444-50. doi: 10.1016/j.drudis.2010.03.013. Epub 2010 Apr 1. PMID: 20362693.
- Jorgensen WL. Efficient drug lead discovery and optimization. Acc Chem Res. 2009;42(6):724-733. doi:10.1021/ar800236t
- Makrynitsa, G.I., Lykouras, M., Spyroulias, G.A. and Matsoukas, M.-T. (2018). In silico Drug Design. In eLS, John Wiley & Sons, Ltd (Ed.). https://doi.org/10.1002/9780470015902.a0028112
- Lu W, Zhang R, Jiang H, Zhang H, Luo C. Computer-Aided Drug Design in Epigenetics. Front Chem. 2018;6:57. Published 2018 Mar 12. doi:10.3389/fchem.2018.00057.